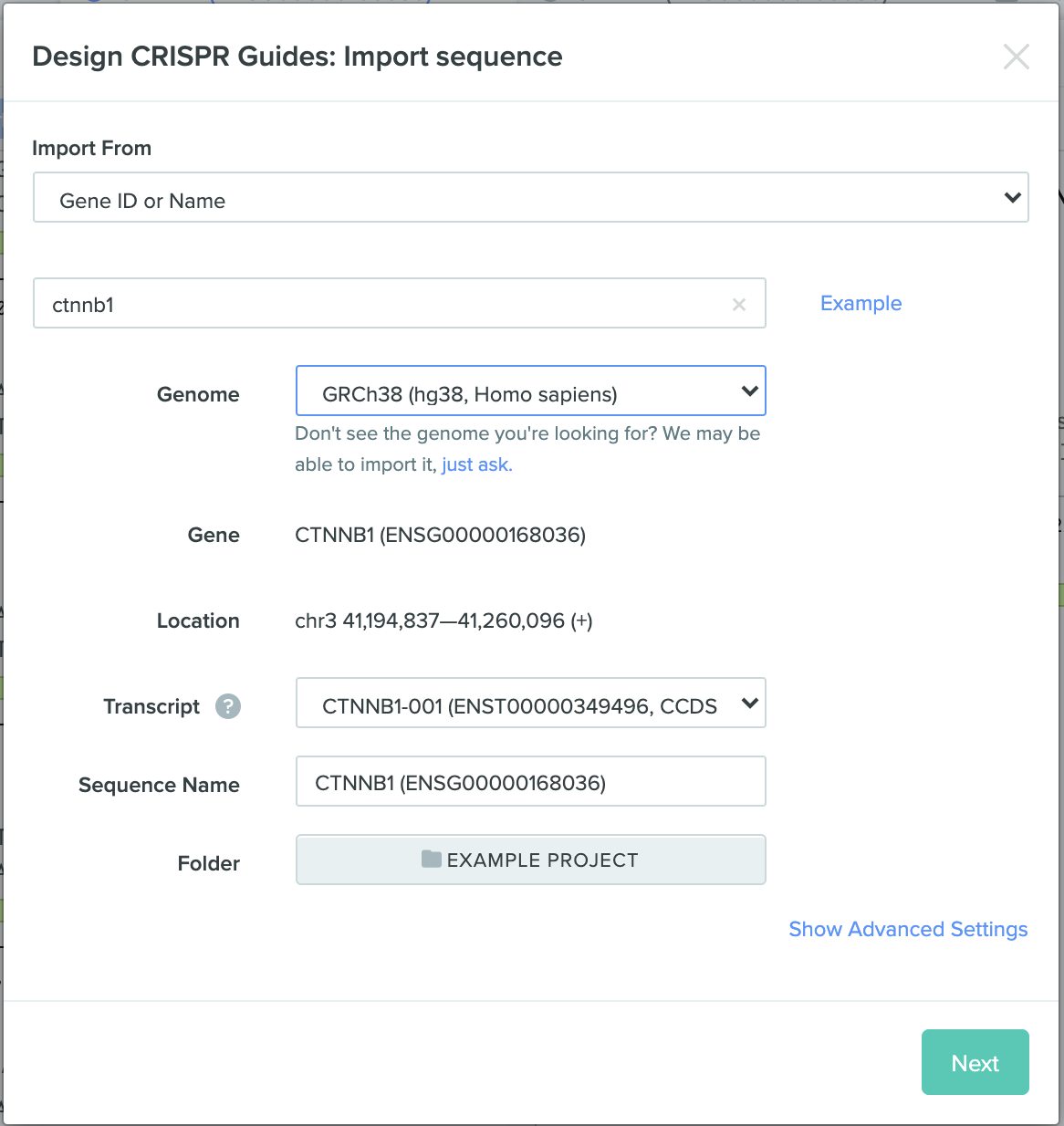
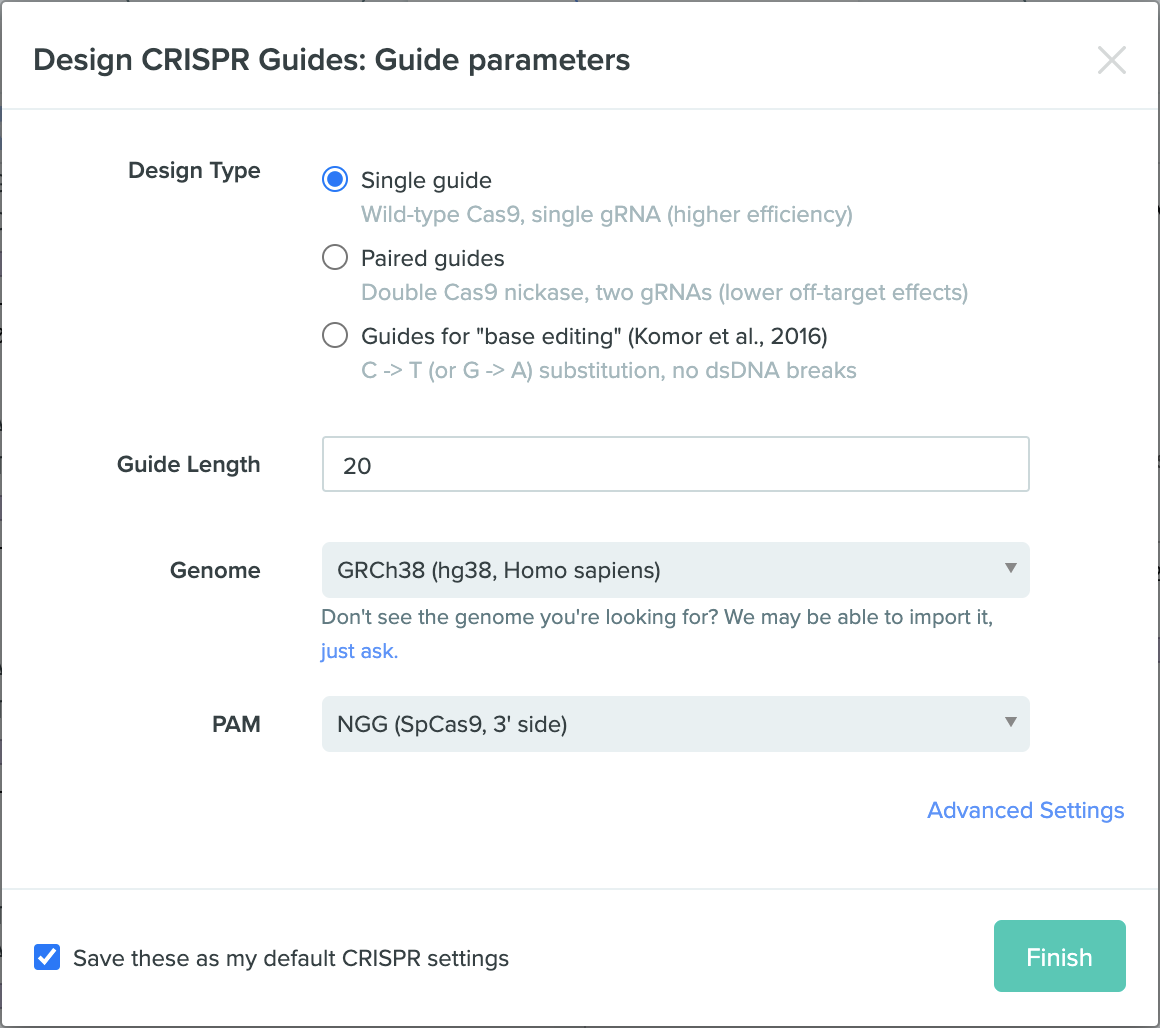
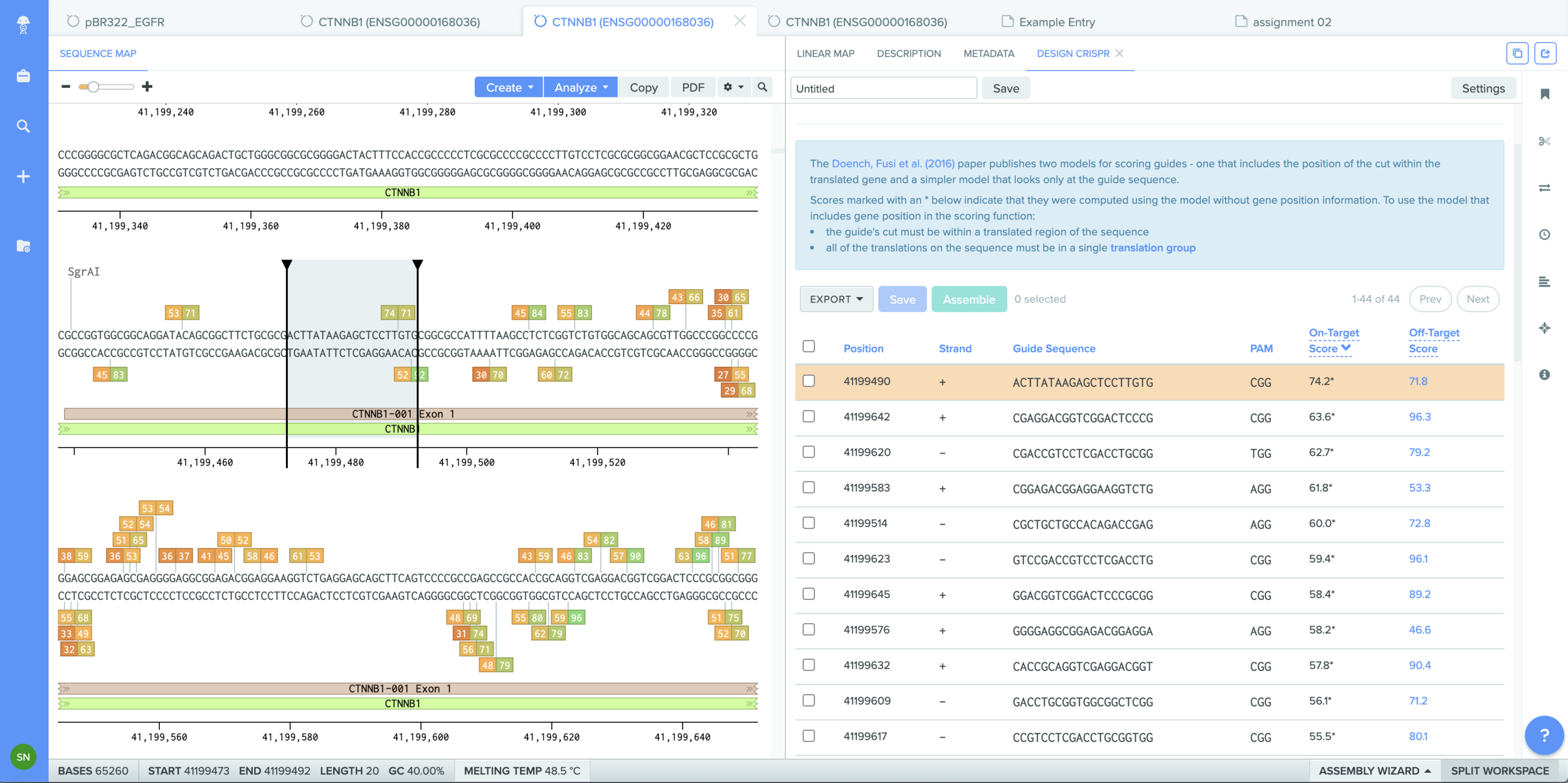
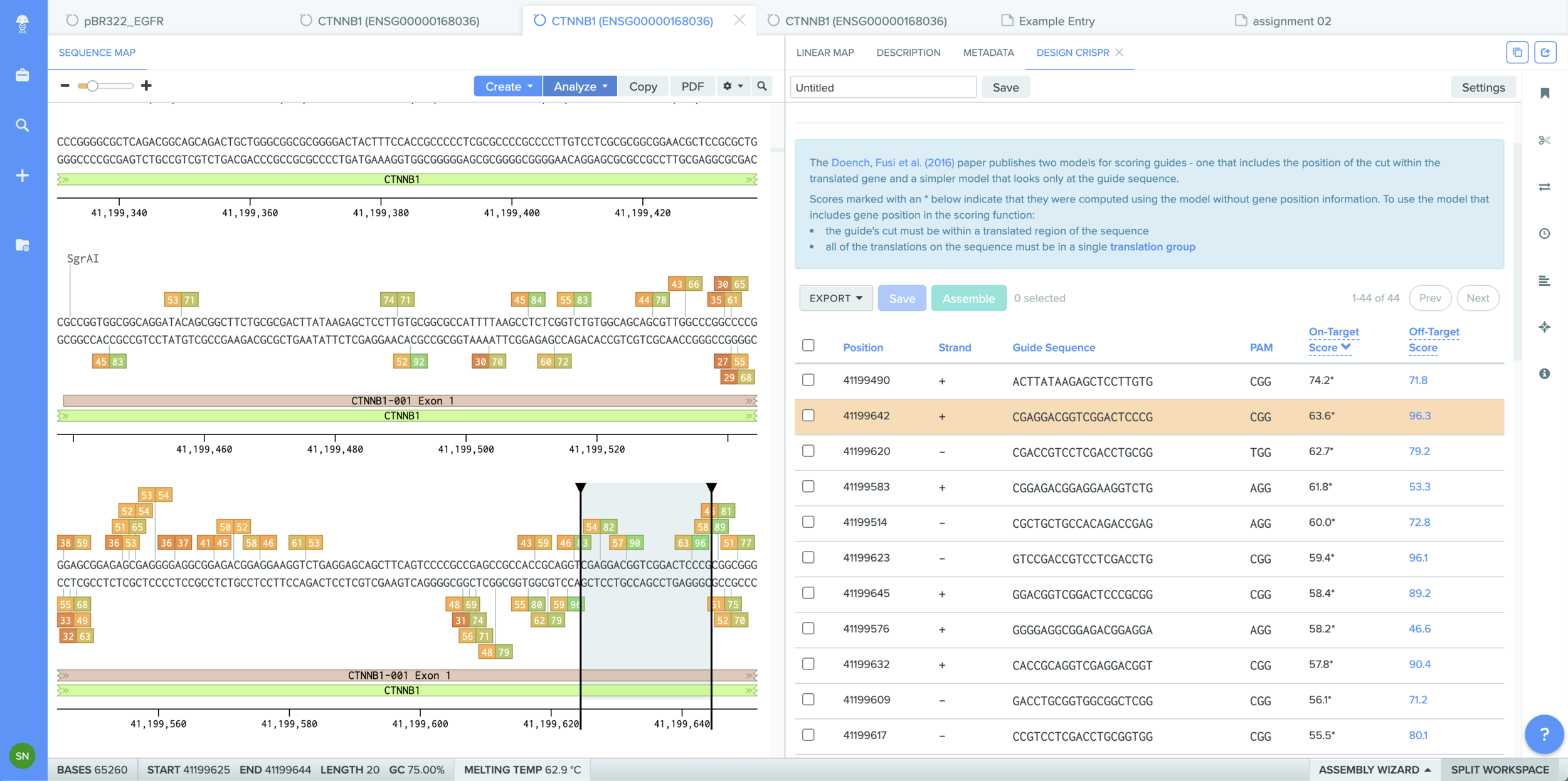
Part I - in silico homework
You will design a useful synthetic minimal cell.
1. Pick a function.
2. Design all components that would need to be part of your synthetic cell.
3. Experimental details
An example solution given below, based on: Lentini, R. et al., 2014. Nat comm, 5, p.4012.
1. Pick a function.
1A What would your synthetic cell do? What is the input and what is the output.
Expand the sensing capacity of bacteria. Input: theophylline (inert to bacteria). Output of the SMC: IPTG. Output of the whole system: GFP produced in bacteria.
Theophyline Aptamer reference: Martini, L. & Mansy, S.S., 2011. Cell-like systems with riboswitch controlled gene expression. Chemical Communications, 47(38), p.10734.
1B Could this function be realized by cell free Tx/Tl alone, without encapsulation?
No. If the IPTG was not encapsulated, it would go into the bacteria without the need of theophylline-induced membrane channel synthesis, thus the synthetic cell actuator would not exist.
1C Could this function be realized by genetically modified natural cell?
Yes, in this particular case: the theophylline aptamer could be incorporated into transformed gene. This lacks the generality though – it is easier to make SMC than modify bacteria, so in this system single bacteria reporter can be used to detect various small molecules.
1D Describe the desired outcome of your synthetic cell operation.
In presence of SMC, bacteria sense theophylline.
2. Design all components that would need to be part of your synthetic cell.
2A What would be the membrane made of?
Phospholipids + cholesterol.
2B What would you encapsulate inside? Enzymes, small molecules.
Cell free Tx/Tl system, IPTG, gene for membrane transporter under the control of theophylline aptamer.
2C Which organism your tx/tl system will come from? is bacterial OK, or do you need mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian).
Bacterial, because of the theophylline riboswitch used as SMC input.
2D How will your synthetic cell communicate with the environment? (hints: are substrates permeable? or do you need to express membrane channel?)
The membrane is permeable to the input molecule (theophylline), the output is IPTG that will cross the membrane via the membrane pore created after theophyline-initiated gene expression.
3. Experimental details
3A List all lipids and genes (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick actual gene)
Lipids: POPC, cholesterol
Enzymes: bacterial cell free tx/tl
Genes: a-hemolysin (aHL) to encapsulate in SMC,
Biological cells: E.coli transformed with GFP under T7 promoter and a lac operator
3B How will you measure the function of your system?
Measure GFP output of the cells, via flow cytometry. Alternatively, use enzymatic reporter, like luciferase, and measure bulk output of the enzyme.
Part II - Experimental homework
For this week’s experimental homework, you get to test different hypotheses about transcription and translation reactions.
Cell-free transcription
You will design a cell-free transcription reaction where the transcription mix produces fluorescent RNA aptamer called Broccoli, see this work by Filonov et al. The RNA is an aptamer that enhances the fluorescence of a small molecule ligand. Fluorescent RNA aptamers. In the transcription reaction, the Broccoli RNA should fold into a conformation which binds a ligand. The ligand--DFHBI--will fluoresce upon binding to the folded Broccoli. Erkin will set-up your designed experiments for you, following the basic recipe below. He will then measure the aptamer fluorescence increase over time using a 96 well plate to check if the mRNA is successfully expressed in the cell-free system. He will share the data with you and you will interpret the results.
This is a good place for you to generate a hypothesis and for Erkin to test your hypothesis. You can try up to 7 condition (perhaps more if the reagents allow it) plus a positive control. You are encouraged to sample different conditions involving the following antibiotics:
-Kanamycin
-Ampicillin
-Rifamycin
-Puromycin
-Vancomycin
- Dactinomycin
-Chloramphenicol
For example, you might want to sample different antibiotics or different concentrations of select few (0.1X, 1X, 10X, etc)
In addition, you might want to play around with the working stock concentrations of the basic kit. For example, transcription reaction includes Magnesium and Potassium salts. Would too little (or too much) of those hurt your reaction? You can also test the effects of different pH (by adding a base or an acids into the reaction) or effects of high energy light exposure (i.e, UV light). You might even ask Erkin to spit into a well. Finally, you can also test temperature effects. The key enzyme in this transcription reaction come from a bacteriophage virus which preys on bacteria that like to reside in our bodies. What would the optimal temperature for this reaction be? How would you test that?
Explain in your webpage the importance of each of the reagents you used to transcribe mRNA using the cell-free system. Think about why including controls in your experiments are important and what your positive control in your experimental design would be. Read about antibiotics in general. What makes a molecule an antibiotic? What are the different modes of action of antibiotics? Interpret the results of your experimental results and compare to your initial hypothesis. What would do differently the next time?
Cell-free transcription and translation
First, Erkin will set up a cell-free protein expression experiment using three different cell-free transcription translation systems as shown below. The template (plasmid) is GFP and its spectrum. Once the reaction is setup, Erkin will incubate the reaction at 30C.
1- PURExpress® In Vitro Protein Synthesis Kit from NEB that is prepared roughly according to the protocol based in this seminal Shimizu et al. paper.
2- BioBits®: Central Dogma Kit that is prepared roughly according to the protocol based in this paper by Huang et al. and stored in a -20oC freezer as a lyophilate for 4 weeks.
3- Homemade cell extract that was stored in a -80oC freezer for less than a week. This system also requires the addition of energy and amino acid mixtures (that are already present in the other two systems above). These mixtures were prepared roughly according to the protocol based in this paper by Sun et al, while the extract was made by sonication according to protocol based on the this paper by Kwan et al.
Think and read about the relationship of transcription to translation. Why are these reactions essential to all life called what they are called? Remind yourself the Central Dogma. Explain in your webpage the importance of each of these reagents used to transcribe mRNA and translate proteins using the cell-free system. Come up at least one advantage of cell-free systems over working with live cells. Bet on which of the three systems will work better. Why did you choose one over the other? Think about how ‘working better’ would look like in this experiment. What would be your output from this experiment? How would you quantify it?
Second, you will again have the chance to generate and test different hypotheses here.
For this, we will mainly utilize BioBits®: Central Dogma Kits Try to design your experiments such that you will be testing a hypothesis. You can try up to 7 condition plus a positive control. Again, you are encouraged to sample different conditions. These include the list of antibiotics and conditions mentioned above. If you decide to test the effects of different reaction components refer to the table above which Erkin will set up using the homemade cell extract.
Think about what your positive control in your experimental design would be. Come up with at least one good negative control. Interpret the results of your experimental results and compare to your initial hypothesis. What would do differently the next time?